Your cart is currently empty!
Capabilities
Protein Analysis
The accuracy, sensitivity and the speed of modern mass spectrometers has been a breakthrough in the field of proteomics. At Allumiqs, our team of experts will perform a full range of analysis from identification and characterization to mapping and quantification and provide our customers with meaningful data.
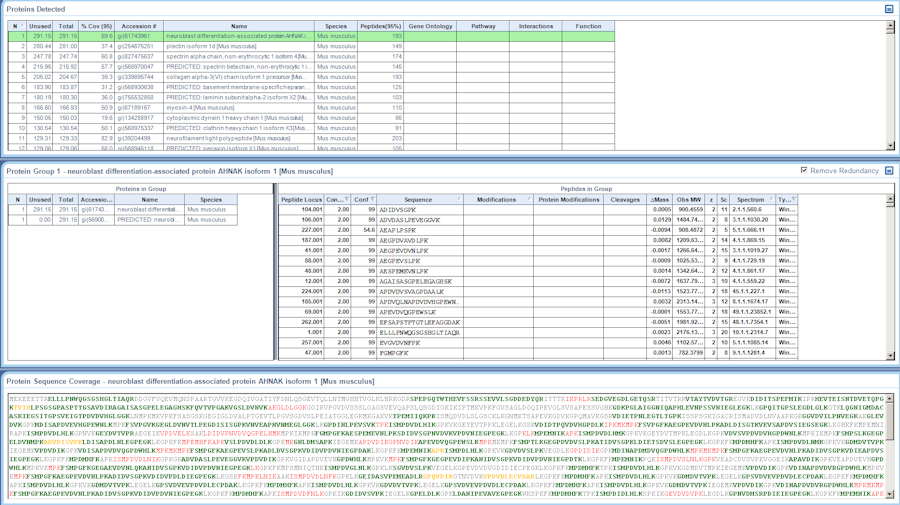
Protein Identification
Identify your protein of interest using LC-MS/MS
We have optimized our protein identification workflow to provide fast and efficient solutions compatible with in-gel digestion of silver nitrate or Coomassie blue stained proteins, as well as for proteins in solution.
In-Gel Protein Identification Workflow
Sample Prep –>
First, we wash the gel pieces with successive dehydration/rehydration cycles in MS-compatible solutions. In a second step, we break intra- and inter-protein S-S bridges using a reducing agent and block the free cysteines by alkylation. The reduced proteins are then digested directly in the gel with a protease that cleaves after known residues. Following digestion, we extract the resulting peptides from the gel by successive rounds of dehydration and sonication. Finally, these peptides are purified using reversed phase SPE (solid phase extraction) and injected in the mass spectrometer.
LC-MS/MS Analysis –>
Prior to analysis on MS, peptides are further separated using a reversed phase liquid chromatography system that is directly connected to the mass spectrometer. Additional separation allows for in-depth analysis in complex samples and increases quantitative and qualitative accuracy. Once inside the MS, peptides are fragmented and signals are recorded by the instrument using an acquisition method named Data Dependent Acquisition (DDA) , also known as Information Dependent Acquisition mode (IDA). The process produces a list of precursor masses (MS) and fragment masses (MS/MS) for further analysis.
Data Analysis & Protein ID
To obtain the identity of the proteins that were in the sample, we load the data in a protein identification software. This software associates the recorded list of masses to a list of peptides and attributes them to one or more proteins. Of note, this process also reports any post-translational modification, chemical modification or amino acid substitution that could be recorded.
Peptide Mapping
Decode protein fingerprints
Peptide mapping is used to confirm the sequence of a protein. After protease digestion, the peptides are separated on a liquid chromatography system and analyzed by mass spectrometry. The protease of choice to perform peptide mapping is trypsin, as its activity is highly predictable and reproducible.
Protein sequence coverage can be improved by using several proteases of different specificity in parallel. By combining the identification results of each protease, we can confirm the sequence of the studied proteins in regions that could not be covered by trypsin because of the intrinsic amino acid sequence of that protein.

Peptide Mapping Workflow
In silico digestion
The first step of confirming the complete amino acid sequence of a protein by mass spectrometry is to cut it into smaller peptides using a protease. The enzyme of choice for many proteomics workflows is trypsin, as it reliably cuts after every lysine and arginine residue (with some exceptions). However, for some regions of your protein, a tryptic digestion may produce peptides that are either too small or too big for proper detection by the machine. This would result in a lack of sequence information for these regions, which is undesirable for a peptide mapping experiment. To prevent this, our experts will first proceed to an in silico digestion of your protein of interest to determine which combination of protease would yield peptides that, when bio-informatically merged, cover 100% of the sequence. We generally achieve full coverage with a combination of two to three different proteases.
Digestion and LC-MS/MS
Once the choice of proteases is set, we will perform the real-life digestion of your protein in the lab. Peptides resulting from each digestion will be analyzed on a short LC-MS/MS gradient using a data-dependent acquisition workflow.
Data Analysis
A protein identification software will first associate the recorded data to a peptide sequence for each protease. This will result in a list of regions that were covered by peptides in each individual protease. Finally, we will pool and align every confirmed peptide from each digestion on the theoretical sequence of the protein to get the final peptide mapping.
Our Omics Capabilities
Proteomics
Get quantitative data for up to 6500 proteins in your samples with our label-free quantitative proteomics workflow.
Lipidomics
Get in-depth profiling of many classes of biologically relevant lipids using our high resolution instruments.
Metabolomics
Sample profiling using either untargeted metabolomics or selected panels of targeted metabolites is the best technique.
Data Analytics
We help customers unlock the value and potential of their data with clearer, deeper insights.
Let’s connect
Get in touch with our experts